Introduction
DoMD (ChemFAST) is a specialized software framework designed to democratize high-throughput molecular simulations. It functions as a “Chemical Compiler”, translating high-level chemical intuition into simulation-ready computational models.
Design Philosophy
The central tenet of ChemFAST is “Chemical Completeness”.
In materials synthesis, chemists manipulate matter through functional groups, monomers, and reaction rules—not by positioning individual atoms in 3D space. Traditional simulation builders, however, often require users to manually define connectivity or rely on rigid templates, creating a disconnect between chemical design and computational implementation.
ChemFAST bridges this gap by adopting a Generalized Topological Representation. It abstracts the complexity of system construction into a generative logic that mirrors laboratory synthesis :
Input: Semantic chemical descriptors (SMILES) and reaction logic (SMARTS).
Process: Automated compilation via stochastic graph transformation and machine learning.
Output: Fully equilibrated, parameterized, and topologically complex molecular systems.
Key Capabilities
ChemFAST is engineered to handle the full complexity of soft matter systems:
Universal Topology Construction: From simple linear chains to hyper-branched polymers and percolated cross-linked networks.
Stereochemical Fidelity: Rigorously preserves chirality (R/S) and cis/trans isomerism during the transition from 2D strings to 3D models.
Automated Parameterization: Eliminates the bottleneck of force field assignment using a hybrid engine that combines database accuracy with Graph Attention Network (GAT) predictions.
High-Throughput Scalibility: Designed for batch processing, enabling the screening of hundreds of materials (e.g., Polyimides) with minimal manual intervention.
Scope and Limitations
While ChemFAST is a versatile toolkit, it is optimized for specific domains of molecular modeling. Users should be aware of the following scope boundaries regarding topological reconstruction and force field applicability.
1. Backmapping and Structural Reconstruction
The S-CGFG (SMILES/SMARTS-driven Coarse-Graining/Fine-Graining) algorithm excels at reconstructing amorphous, flexible matter.
- Supported Systems:
Synthetic Polymers: Linear, star, and ring polymers; random/block copolymers.
Complex Networks: Epoxy resins, hydrogels, and solid polymer electrolytes (SPEs) formed via in-situ polymerization.
Stereochemistry: The builder strictly enforces local stereochemical constraints (e.g., tacticity in polypropylene, cis/trans bonds in polyisoprene) defined in the input SMILES.
- Limitations (Biomacromolecules):
ChemFAST is NOT a protein folding engine. The ab initio prediction of tertiary structures for complex biological macromolecules (Proteins, DNA, RNA) is beyond the scope of this toolkit. * Solution: We employ a Rigid-Body Protocol. Users must provide experimentally resolved structures (e.g., PDB files) or high-quality predictions (e.g., from AlphaFold). ChemFAST treats these as fixed topological scaffolds, building the flexible synthetic matrix around them to construct bio-hybrid assemblies (e.g., Core-Shell nanoparticles).
2. Force Field Assignment
The assignment engine is built upon the OPLS-AA framework.
- Target Domain:
Materials Science: Organic molecules, synthetic polymers, and electrolytes.
The hybrid database/ML approach is extensively validated for these systems, covering partial charges and bonded parameters (via GATs) for a vast chemical space.
- Limitations:
Bio-specific Potentials: ChemFAST is currently not optimized for specialized biological force fields such as OPLS-AA/M, AMBER or CHARMM. Users working strictly on biomolecular dynamics may need to validate the OPLS parameters or use the User-Defined Force Field API to inject external parameters.
Inorganic Systems: For inorganic systems,ChemFAST relies on interface-specific parameters while standard OPLS-AA force field may failed.
DoMD Workflow
DoMD functions as a “Chemical Compiler” for molecular dynamics. Instead of manually drawing topologies or patching residue files through a GUI, you provide high-level chemical definitions via Python scripts, and DoMD compiles them into simulation-ready files.
The overall workflow consists of three major stages:
Chemical Definition: You define what molecules exist using SMILES strings and how they connect using SMARTS reaction templates.
S-CGFG (SMILES/SMARTS-driven Coarse-Graining/Fine-Graining): ChemFAST generates a relaxed system using efficient Coarse-Grained (CG) pre-equilibration and then reconstructs the All-Atom (AA) details from the CG coordinates.
Force Field Assignment: The reconstructed all-atom system is typed using a hybrid approach that combines database lookups with Machine Learning (GAT) predictions to generate the final topology.
Flowchart of a Typical Simulation
A typical simulation workflow with DoMD is illustrated below. This flowchart represents the transformation from raw chemical input strings to a production-ready GROMACS run, using Polyimide synthesis as an example.
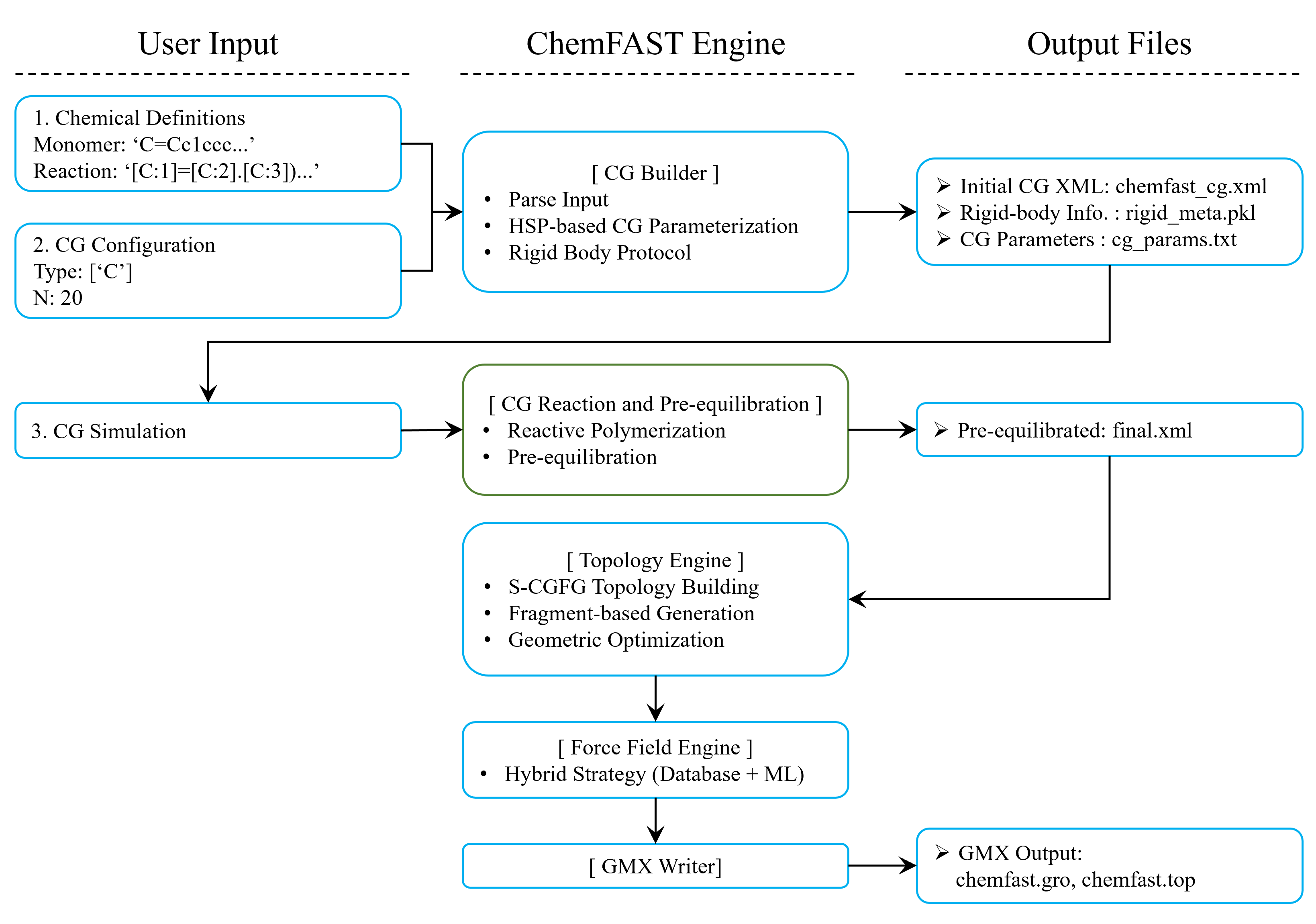
Important Files
Here is an overview of the most important file types you will encounter when working with DoMD.
The Control Script (.py)
ChemFAST is controlled entirely via Python scripts. This script contains your Chemical Definitions (SMILES/SMARTS), system composition rules (e.g., number of chains, degree of polymerization), and calls to the high-level domd_tools API. This approach allows for powerful, programmable logic, such as using loops to generate complex polymer brush architectures.
Chemical Definition Strings (SMILES/SMARTS)
ChemFAST use SMILES/SMARTS (or PDB files) to define initial chemistry.
* SMILES: Defines the atomic connectivity and structure of your monomers (e.g., CCO for Ethanol).
* SMARTS: Defines the reaction rules for polymerization, crosslinking, or connecting fragments (e.g., [C:1]=[C:2] >> [C:1]-[C:2]).
* Note: Correct atom map indices within SMARTS are crucial for defining regio-specificity (e.g., ensuring Head-to-Tail connectivity in vinyl polymers).
Coarse-Grained Configuration (.xml)
Generated by the create_cg_system tool. This is a PyGAMD/HOOMD-blue (v 1.3.3) compatible XML file containing the Coarse-Grained representation of your system.
The generation of these CG topologies proceeds via three distinct pathways:
In situ Polymerization: Used during reactive CG polymerization simulations, where bond formation events are explicitly logged by the simulation engine.
Algorithmic construction: Uses Self-Avoiding Random Walks (SARW) with step length and excluded volume derived from a conformational analysis of representative oligomers generated via SMILES/SMARTS.
Post-hoc reconstruction: Uses a Breadth-First Search (BFS) algorithm to infer connectivity logic from pre-assembled configurations.
Crucially, for intricate topological architectures such as cross-linked networks, the in situ polymerization method is required to ensure the physical validity of the network structure. This file is primarily used for the Pre-equilibration step to relax polymer chains efficiently before backmapping.
Rigid Body Metadata (.pkl)
If your system involves rigid biomolecules (e.g., Proteins, DNA) imported from external PDB files, DoMD generates a pickle file (e.g., rigid_meta.pkl). This file links the coarse-grained beads back to the original atomic structure in the PDB file, ensuring the tertiary structure and orientation are correctly preserved during the backmapping process.
GROMACS Topology (.top) and Structure (.gro)
These are the final output files generated by DoMD’s exporter.
* .gro: Contains the equilibrated all-atom coordinates reconstructed from the CG simulation.
* .top: Contains the complete system topology including atom types, charges (assigned via ML/DB), bonds, angles, and dihedrals (OPLS-AA). These files are ready to be processed by gmx grompp.
Tutorial Material
Several tutorials are available in the TUTORIALS that cover specific use cases, ranging from simple linear polymer melts to complex bio-hybrid assemblies and reactive networks. We recommend starting with the basic Linear Polymer tutorial to understand the fundamental pipeline before moving to more advanced examples.
The GitHub repository includes a comprehensive, step-by-step tutorial for a linear polyimide melt, covering the complete workflow from chemical definition to final GROMACS files. To help users understand the underlying algorithms and data flow, we also supply individual test scripts for specific modules (such as CG topology generation and backmapping).
For extended applications, please download the examples package from the GitHub Releases, which contains tutorials for:
Linear Polymers: Generating linear polyimide melts with controlled tacticity and molecular weight distributions.
Brush Polymers: Building densely grafted polymer brushes with tunable grafting densities and side-chain lengths.
Cross-linked Networks: Modeling SPEs network formed via in-situ polymerization, including percolated network structures.
Bio-hybrid Assemblies: Constructing core-shell nanoparticles (rigid protein cores with flexible polymer shells) using experimental PDB structures.
Membrane Systems: Backmapping of lipid bilayers and AuNPs complexes.