Lipid Membranes & Protein-Loaded NPs (examples/aunps)
This tutorial demonstrates the Resolution-Agnostic capability of DoMD. We will reconstruct a highly complex system typical of Martini-level coarse-graining: a lipid membrane interacting with PEGylated Gold Nanoparticles (AuNPs) and protein-loaded Lipid Nanoparticles (LNPs).
Key concepts introduced here:
Martini-to-All-Atom Mapping: Mapping coarse-grained beads (representing chemical fragments like Phosphates, Glycerol, Alkyl tails) back to atomic resolution.
“Artificial” Reaction Templates: Using dummy atoms (Fluorine) as positioning markers to define connectivity between fragments.
Complex Heterogeneity: Simultaneously handling metals, polymers, lipids, and peptides in a single workflow.
Important
The “Artificial Chemistry” Technique
In this tutorial, the chemical reactions defined in reaction_template are NOT physically real reactions. They are topological instructions used to stitch fragments together.
Notice the frequent use of Fluorine (F) atoms in the SMILES strings (e.g., PEO = 'CCOF'). Here, ‘F’ acts as a “Connector Port” or “Positioning Marker”.
Logic: The SMARTS pattern
[C:1][F:2] + [C:3] >> [C:1][C:3]tells DoMD to bond the carbons and delete the ‘F’ atom.Purpose: This ensures that when fragments are assembled, they strictly maintain the correct orientation and valency, effectively “snapping” together like Lego blocks.
Step 1: Fragment Definitions (The Building Blocks)
We define the SMILES for every component. Note that these are not just whole molecules, but fragments corresponding to CG beads (e.g., Martini beads).
Lipid Parts: Headgroups (NC3, PO4), Glycerol (GL1, GL2), Tails (C1A, D2A…).
Peptide Residues: Specific amino acid fragments (LYS, TRP, VAL…).
Nanoparticle: Gold (Au) and PEGylating agents (PEO).
from domd_tools import create_cg_system, build_aa_topology, assign_ff_parameters, get_gmx
from misc.logger import logger
logger.setLevel('INFO')
# --- 1. Define Components (SMILES) ---
# PEO chain and Au
PEO = 'OCCO'
Au = '[Au]'
# Peptide Residues (Targeting a specific sequence)
LYS1 = 'NCCCC[C@@H](N)C(=O)O'
VAL1 = 'CC(C)[C@@H](N)C(=O)O'
TRP1 = 'c1cccc2c(C[C@@H](N)C(=O)O)cnc2c1'
# ... (Refer to the full script for all residues) ...
# Lipid Fragments (Martini mapping logic)
NC3 = 'C[N+](C)(C)C' # Choline head
PO4 = 'OP(=O)([O-])OC' # Phosphate
GL1 = 'FC[C@@H](OC(=O)CC)CO' # Glycerol backbone (with F marker)
GL2 = 'C(=O)CC'
GL0 = 'OCCO'
C1A = 'CCCF' # Saturated tail fragment
D2A = 'CCC=CCC' # Unsaturated tail fragment
# ... (Refer to the full script for all lipid parts) ...
Step 2: All-Atom Connectivity Definitions (The Reaction Template)
This is the core of the resolution-agnostic approach. We define how these fragments connect to form the full molecules (Lipids, Peptides, PEO-Au).
Observe the SMARTS pattern: [CH3:1].[C:2][F:3]>>[CH2:1][C:2].[F:3].
This translates to: “Take a Methyl group, connect it to the Carbon holding the Fluorine, and eject the Fluorine.”
# --- 2. Define Connectivity Rules ---
reaction_template = {
# --- Polymerization & Grafting ---
'a': { # PEO-PEO chain growth
'cg_reactant_list': [('PEO', 'PEO')],
'smarts': '[C:1][O:2].[O:3][C:4]>>[C:1][O:2][C:4].[O:3]',
'prod_idx': [0]
},
'b': { # Grafting PEO to Gold Surface
'cg_reactant_list': [('Au', 'PEO')],
'smarts': '[Au:1].[OH1:2][C:3]>>[Au:1][O:2][C:3]',
'prod_idx': [0]
},
# --- Lipid Assembly (Stitching Head-Glycerol-Tails) ---
'NC3-PO4': {
'cg_reactant_list': [('NC3', 'PO4')],
'smarts': '[C:1].[C:2]>>[C:1][C:2]',
'prod_idx': [0]
},
'PO4-GL1': {
'cg_reactant_list': [('PO4', 'GL1')],
'smarts': '[O:1].[C@@H:2][C:3][F:4]>>[O:1][C:3][C@@H:2].[F:4]',
'prod_idx': [0]
},
'GL1-C1A': { # Connecting Tail to Glycerol
'cg_reactant_list': [('GL1', 'C1A')],
'smarts': '[CH3:1].[C:2][F:3]>>[CH2:1][C:2].[F:3]',
'prod_idx': [0]
},
# --- Peptide Synthesis (Peptide Bonds) ---
'VAL1-TRP1': {
'cg_reactant_list': [('VAL1', 'TRP1')],
'smarts': '[NH2:1][C@@H1:2].[C:3](=[O:4])[OH1:5]>>[C:3](=[O:4])[NH1:1][C@@H1:2].[OH2:5]',
'prod_idx': [0]
},
# ... (Additional peptide bonds for LYS, TRP, etc.) ...
}
Step 3: All-Atom Topology Building
We load the fragments into the mols dictionary. Then, we perform backmapping on a pre-existing CG snapshot (cg/lipidmem_with_AuNP.xml). This snapshot represents the equilibrated state of the Martini-level simulation.
# --- 3. Register Molecules ---
# (Full dictionary from the script)
mols = {
'PEO': {'smiles': PEO, 'file': None, 'is_rigid':False},
'Au': {'smiles': Au, 'file': None, 'is_rigid':False},
'LYS1': {'smiles': LYS1, 'file': None, 'is_rigid':False},
# ... (Full list of molecules) ...
'C1A': {'smiles': C1A, 'file': None, 'is_rigid':False},
'W': {'smiles': 'O', 'file': None, 'is_rigid':False},
}
# --- 4. Backmapping ---
# Input: A Martini-scale CG configuration
xmlfile = 'cg/lipidmem_with_AuNP.xml'
# Reconstruct AA Topology
# large=50 ensures spatial decomposition is used for large membrane/NP structures
confs, mol_graphs, box = build_aa_topology(
mols,
reaction_template,
xmlfile,
large=50
)
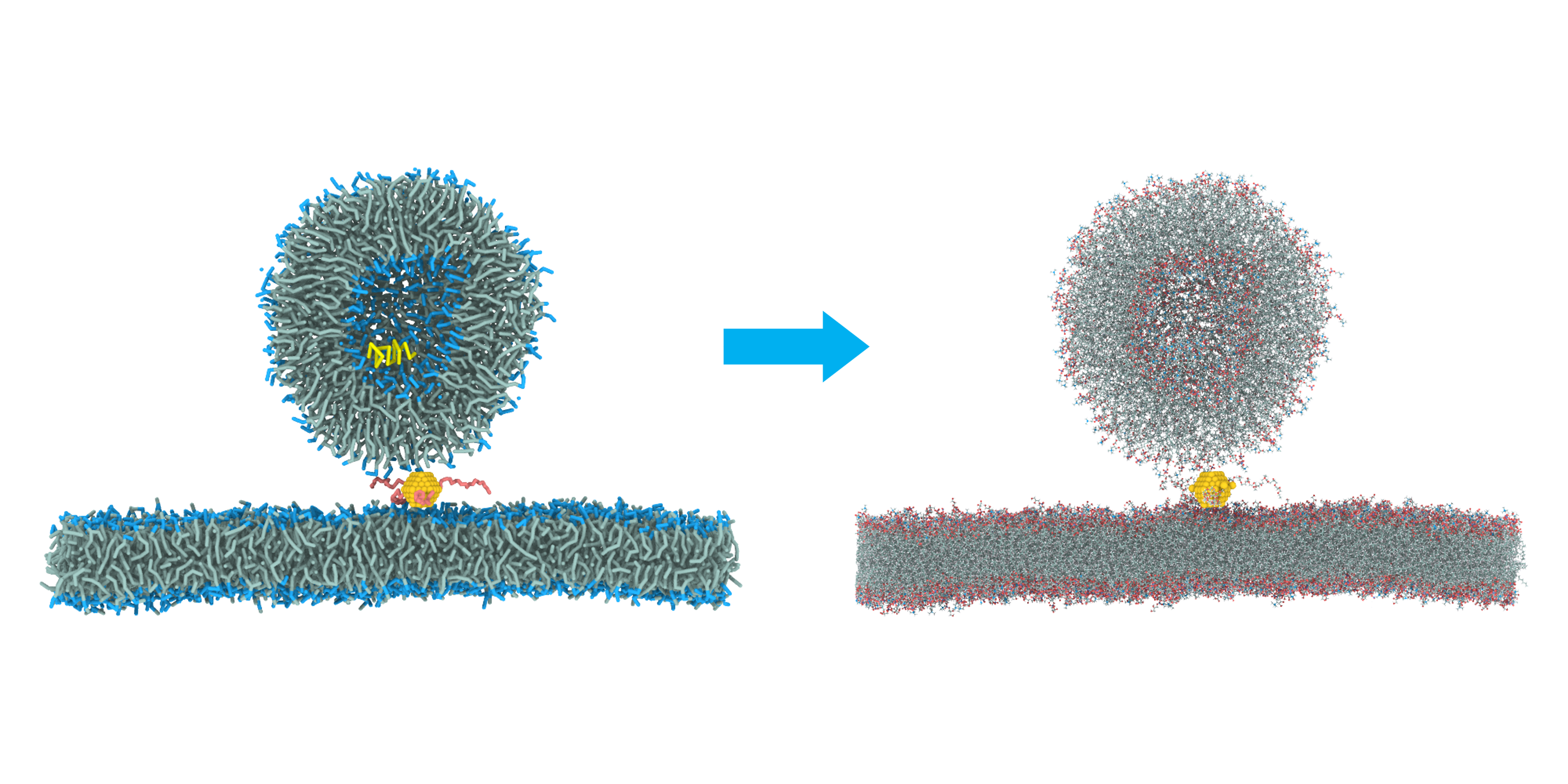
Figure 1: Visualization of the backmapping process. DoMD replaces the coarse-grained beads (left) with the defined chemical fragments (right), removing the ‘F’ connectors to form continuous molecules.
Step 4: ML Force Field & Output
Given the complexity and heterogeneity of the system (Lipids + Gold + Peptides), we employ the Machine Learning (ML) force field strategy. This ensures that parameters for the specific lipid fragments and the gold interface are handled consistently without manual atom-typing.
# --- 5. Force Field Assignment ---
# Use Pure ML strategy for all components except single atoms (e.g., Au, ions) which can use TPL
strategies = []
for mol_graph in mol_graphs:
if mol_graph.number_of_nodes() == 1:
strategies.append('tpl')
else:
strategies.append('ml')
ffs = assign_ff_parameters(mol_graphs, strategies=strategies)
# --- 6. Export ---
get_gmx(mol_graphs, confs, ffs, box)
Final Result: DoMD successfully translates the “beads” of the Martini-like input into fully connected, chemically accurate all-atom structures of:
A Phospholipid Bilayer.
Gold Nanoparticles with grafted PEG chains.
Peptide/Protein chains embedded or interacting with the system.