Custom Force Field Construction
ChemFAST is designed with an Open Force Field Architecture. While it comes pre-packaged with a comprehensive OPLS-AA database, research often requires specialized parameters (e.g., for novel ionic liquids, transition metal complexes) or entirely different force field families (e.g., AMBER/GAFF).
This section details the workflow for constructing or extending the force field SQL databases using the WL-Hash Subgraph Matching engine.
1. The Underlying Mechanism: WL-Hashing
Traditional force field implementations often hardcode “atom types” into the software logic. ChemFAST decouples data from logic using Weisfeiler-Lehman (WL) Graph Hashing.
The Concept: The engine computes a unique cryptographic hash for every atom based on its local topological environment (element, hybridization, neighbors up to \(N\) hops).
The Database: The SQL database acts as a key-value store:
Key (WL-Hash) -> Value (Charge, Sigma, Epsilon) "a1b2c3d4..." -> {q: -0.2, sigma: 0.35, epsilon: 0.066}The Benefit: To add a new molecule, you do not need to modify source code. You simply compute the hashes for your new molecule and inject the corresponding parameters into the database file.
2. Unified Database Management
ChemFAST provides a unified utility function update_data_to_db to handle both updating existing databases and creating new ones.
The logic is controlled via the create switch, but the system is smart enough to auto-detect context:
If the target database file exists, it defaults to Update Mode (appending new hashes).
If the target database file is missing, it automatically switches to Create Mode (initializing schema and populating).
Tool Location: domd_database/manage_db.py (or specific script location)
3. Usage Workflow
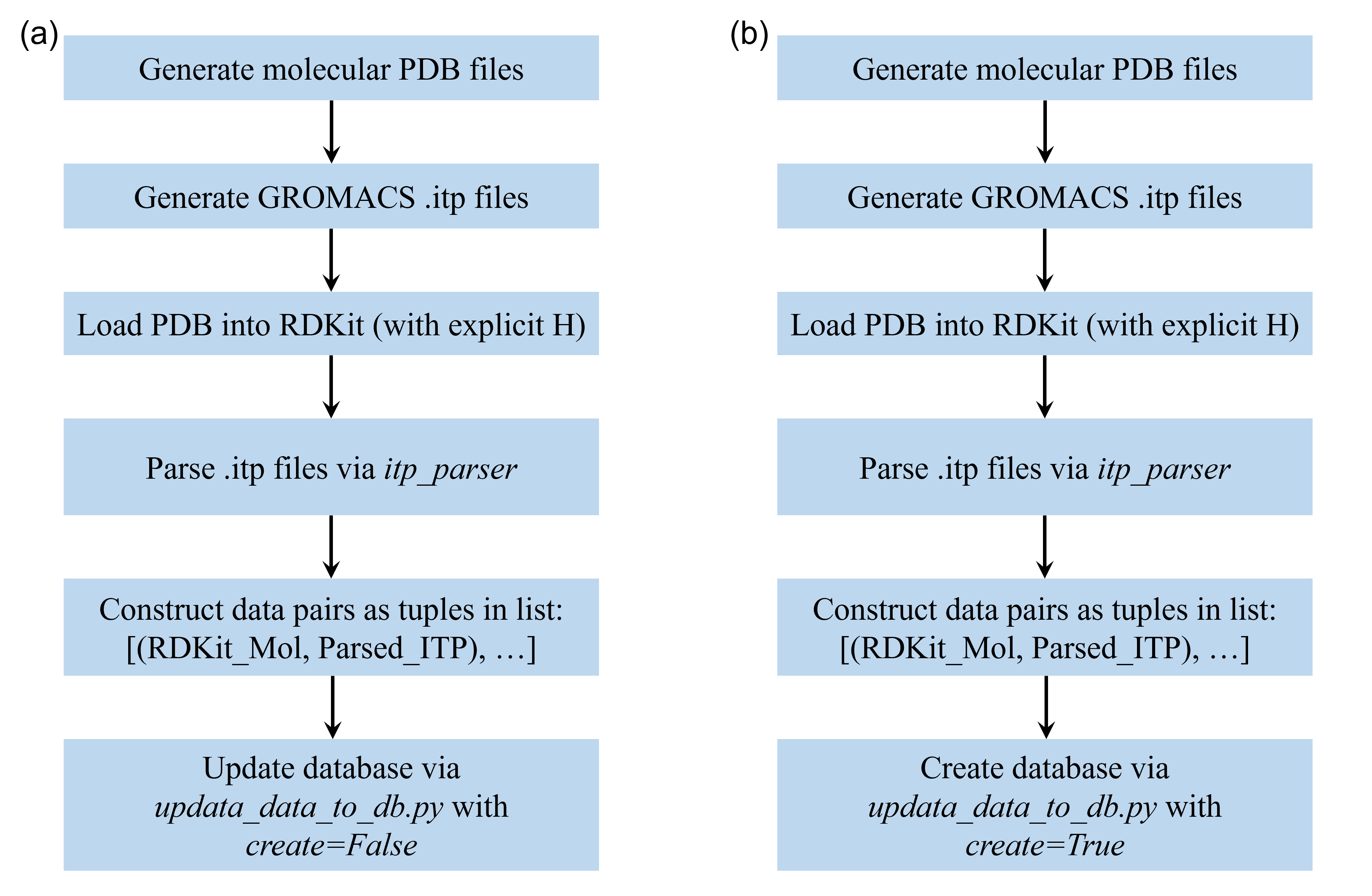
Whether you are adding a single ionic liquid to OPLS-AA or building a fresh GAFF database from scratch, the workflow is identical.
Step 1: Input Generation Prepare the “Ground Truth” files using external tools (e.g., LigParGen, Antechamber/Acpype, or QM fitting):
structure.pdb: Defines connectivity and elements. Must contain explicit hydrogens.topology.itp: Contains the force field parameters (charges, bonds, etc.) in GROMACS format.
Step 2: Execution via Python Script
You can use the update_data_to_db function to batch process multiple molecules.
from domd_database.manage_db import update_data_to_db
# Define your input lists (Order must match!)
my_pdbs = ['mol1.pdb', 'mol2.pdb', 'ionic_liquid.pdb']
my_itps = ['mol1.itp', 'mol2.itp', 'ionic_liquid.itp']
# --- Scenario A: Extending the OPLS Database ---
# This will check 'opls.db'. If it exists, it updates it, else it creates it.
update_data_to_db(
db='opls',
db_path='opls.db',
itps=my_itps,
pdbs=my_pdbs
)
# --- Scenario B: Creating a New GAFF Database ---
# If 'my_custom_gaff.db' does not exist, create=True is automatically inferred.
update_data_to_db(
db='gaff',
db_path='my_custom_gaff.db',
itps=my_itps,
pdbs=my_pdbs
# create=True <-- Optional, auto-detected if file is missing
)
Parameters Explained:
db: The force field type ('opls'or'gaff'). This determines which parser to use (LigParGen vs. Acpype).db_path: The filename of the SQL database.create: (Bool) Explicitly force creation of a new DB. IfFalsebut the file is missing, the code overrides it toTrue.
4. Usage
Once the script finishes:
WL-Hashes are computed for every unique atom environment in your PDBs.
Parameters are extracted from the ITPs.
The SQL Database is populated.
You can now use this custom or updated database in your simulation scripts. Instead of using the high-level wrapper, you explicitly instantiate the Force Field class (e.g., GAFF) and pass your database object directly to it.
Here is a complete example of how to deploy it:
from domd_forcefield.gaff.gaff import GAFF
from domd_forcefield.gaff.gaff_db import gaff_db
from domd_tools import get_gmx
import pickle
import numpy as np
from rdkit import Chem
from rdkit.Chem import AllChem
# 1. Prepare your molecule
mol = Chem.MolFromSmiles('[H]c1nc(C(=O)c2c([H])c([H])c(OC([H])([H])[H])c([H])c2[H])c(Cl)c([H])c1C(F)(F)F')
molh = Chem.AddHs(mol)
AllChem.EmbedMolecule(molh)
AllChem.UFFOptimizeMolecule(molh)
# Set necessary properties for exporting
for a in molh.GetAtoms():
a.SetIntProp('res_id', 0)
a.SetIntProp('global_res_id', 0)
a.SetProp('res_name', 'MOL')
confs = [molh.GetConformer(0).GetPositions()]
box = np.array([10, 10, 10]) * 10 # Angstroms
# 2. Instantiate the Force Field with your specific database
ff = GAFF(database=gaff_db)
# 3. Parameterize the molecule
ff.parameterize(molh, custom_rules='none', boss_radius=2)
# 4. Extract parameters and export
ffs = [ff.all_params]
mols = [molh]
pickle.dump(ffs, open('meta_ffs.pkl', 'wb'))
get_gmx(mols, confs, ffs, box)